AI in Biology: AI Meets Intrinsically Disordered Proteins
Protein folding — the process by which a protein arrives at its functional shape — is one of the most complex unsolved problems in biologyThe Atlantic Magazine’s creative marketing studio Re:think now dedicates an entire publication to the changes AI has wrought and what may come next. The magazine, titled Dialogues: On AI, Society, and What Comes Next, is produced with tech giant Google. As you might expect in such a partnership, it leans heavily into extolling the virtues of AI rather than cautioning readers about its limitations.
In the introduction to the latest edition — Dialogues is published quarterly — the editors point out that “[m]uch of the current conversation around the rise of artificial intelligence can be categorized in one of two ways: uncritical optimism or dystopian fear.” True enough. And, we suspect, also true is Dialogues’ take: “The truth tends to land somewhere in the middle — and the truth is much more interesting.”
The problem for Re:think and Google is that the essays in the new publication tend to be far from the middle. The word “cheerleading” comes to mind.
Take this edition’s feature, “The AI-Powered Future of Drug Discovery.” I’m not a biologist, much less a structural one, but I have an interest in the intersection of AI and biology as a computer scientist specializing in natural language processing (NLP), an important subfield of AI. A friend, colleague, and former professor I know has dedicated his professional life to understanding the limits of reductive mechanisms in biology. He originally turned me toward the bold claims made on behalf of deep neural network systems predicting how proteins fold. I’ve been hooked ever since.

So, when the new edition of Dialogues advertised itself in my print version of The Atlantic, I read the “truth is somewhere in the middle” salvo and expected to get a nuanced view of AI’s successes and failures on the notoriously complicated protein folding problem.
I didn’t.
Reading Dialogues reminded me of something Meredith Broussard discusses in her book Artificial Unintelligence: How Computers Misunderstand the World (MIT Press 2018). Broussard argues that AI is often hyped as a miracle solution — a tool that will replace human expertise, automate complexity, and deliver results with scientific precision. In reality, AI is deeply constrained by the limits of the data it has been trained on, and nowhere is this more evident than in the world of protein folding prediction.
Despite what the AI hype cycle might lead us to believe, the hard problems in biology have not suddenly become easy. Dialogues has more than trumpeted the positives and hyped the future possibilities—it has largely ignored the serious limitations.
So let’s take a look at reality.
The Protein Folding Problem
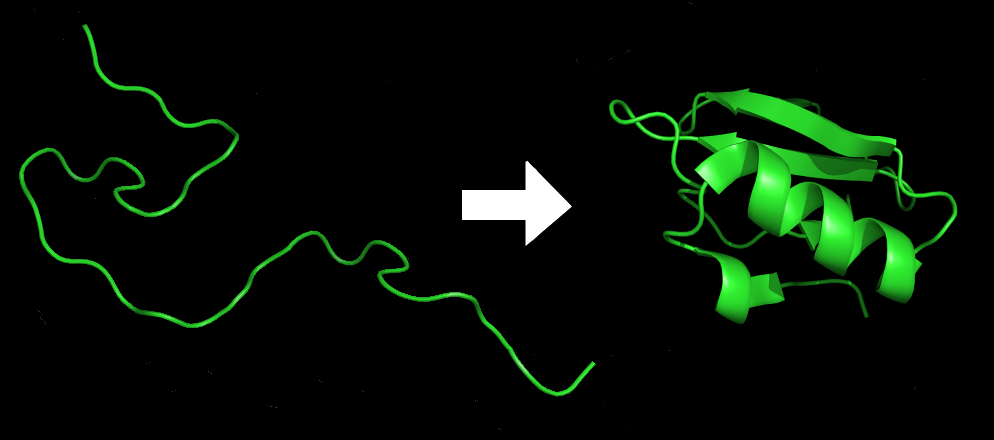
Proteins are the fundamental building blocks of life, carrying out a vast range of functions in the cell. Their functions depend critically on their three-dimensional structures, which arise from the way a sequence of amino acids folds. The process by which a protein arrives at its functional shape is known as protein folding, and for decades, it has been one of the most complex unsolved problems in biology. Scientists have sought to predict protein structures from their amino acid sequences—what is known as “the protein folding problem.”
For years, experimental techniques such as X-ray crystallography and cryo-electron microscopy (cryo-EM) were the only reliable ways to determine a protein’s structure. These methods, while highly accurate, are also slow, expensive, and labor-intensive. The technologies of the lab, we might say, have been pivotal in the success of modern biology but also have forced scientists and technicians into a laborious ritual that in some cases takes years to complete. Still, the methods are somewhat ingenious.
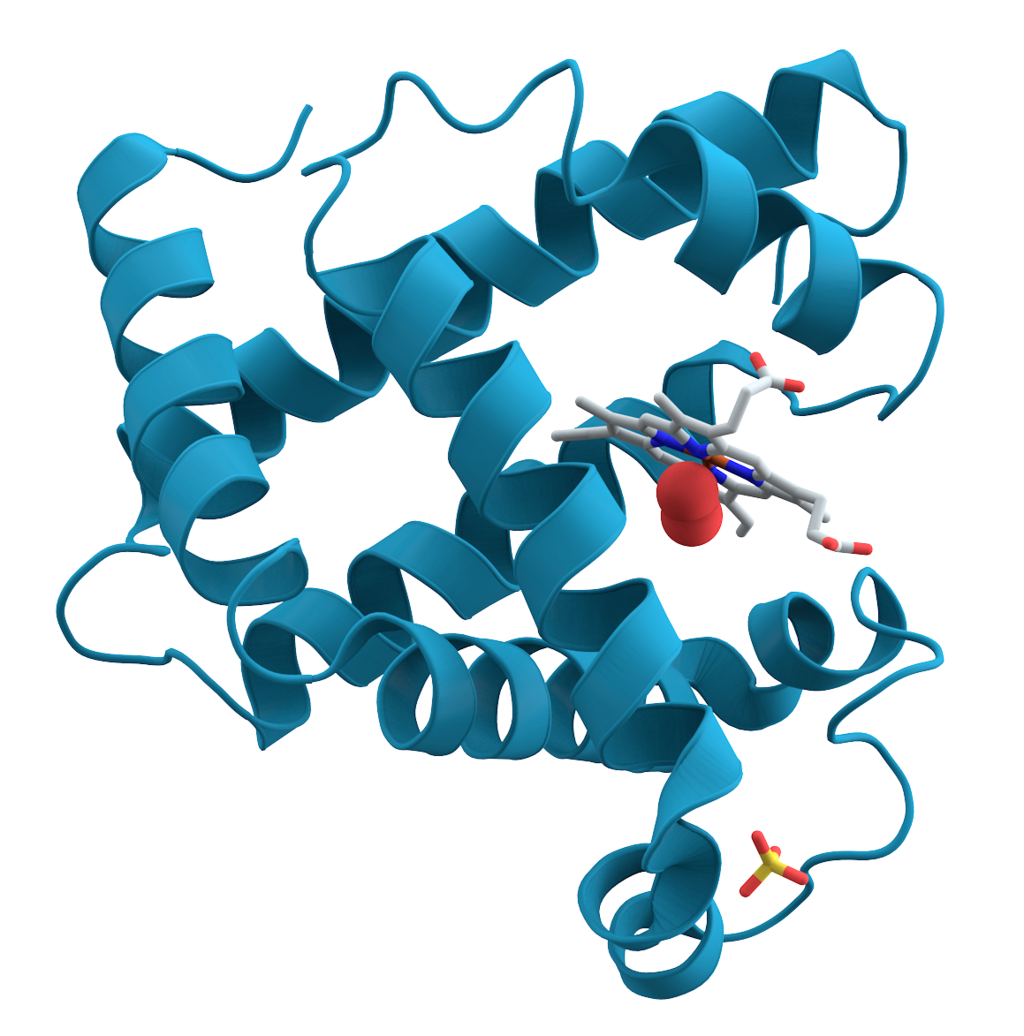
showing alpha helices.
Such proteins are long, linear molecules with
thousands of atoms; yet the relative position
of each atom has been determined with
sub-atomic resolution by X-ray crystallography.
Take X-ray crystallography, for example. Here the target protein is purified and coaxed into forming a crystalline structure, an arduous process that can take weeks, months, or even years for some proteins. And not all proteins are cooperative — some simply refuse to crystallize, rendering them nearly invisible to this technique. But for those that do, the results can be remarkable. Once crystallized, the sample is bombarded with high-energy X-rays, and the resulting diffraction patterns are analyzed to reconstruct the molecule’s atomic arrangement.
X-ray crystallography was famously used in the 1950s to help uncover the double-helix structure of DNA, an achievement that fundamentally reshaped our understanding of genetics. Since then, crystallography has illuminated the structures of countless biological molecules, from enzymes to viral proteins, laying the foundation for modern molecular biology.
Cryo-electron microscopy (cryo-EM) is equally laborious — with results that can also be impressive. Rather than forcing a protein into a crystalline lattice, cryo-EM involves rapidly freezing proteins in solution at extremely low temperatures — a process that locks them in place while preserving their natural shape. The sample is then bombarded with an electron beam, and thousands of two-dimensional images are computationally combined to generate a three-dimensional model. Unlike X-ray crystallography, cryo-EM does not require crystallization, making it invaluable for studying proteins that resist the rigid constraints of crystallography or exist in multiple conformations.
However, cryo-EM is not without its limitations. While its resolution has improved dramatically — earning its creator a Nobel Prize in Chemistry in 2017 — the method remains technically challenging and expensive. The images produced are averages of thousands of molecular snapshots, meaning that for highly flexible proteins, the final structure can be blurry or incomplete. Further, some classes of proteins still prove elusive, and interpretation of cryo-EM data requires significant expertise, making it less of an instant solution than enthusiasts might suggest.
To be sure, X-ray crystallography and cryo-EM are indispensable tools in the structural biologist’s toolbox. They have been part and parcel of cell biology for decades, enabling researchers to map the architecture of life’s molecular machinery with stunning precision. The double-helix structure of DNA, the atomic details of hemoglobin, the virus structures that help design vaccines — all of these discoveries have depended on these methods. And yet, despite their power, both techniques come with limitations. X-ray crystallography requires proteins to be coaxed into a crystalline form, a process that can take months or even years, and not all proteins crystallize well. Cryo-EM, while more forgiving, still demands painstaking effort, costly equipment, and expert interpretation to resolve complex molecular assemblies.
More recently, faster computational methods have taken center stage — bringing with them as much confusion as excitement. The successes of AI-driven approaches like AlphaFold, developed by Google’s DeepMind, have been hailed as a revolution in protein folding prediction. AI proponents and publications like Dialogues have framed AlphaFold as a turning point, a breakthrough that could dramatically accelerate protein structure discovery, shrinking time scales that were once measured in years down to mere hours.
But, as is so often the case with AI narratives, the reality is more complicated.
Note: Erik J. Larson writes the Substack Colligo.
Here are the rest of the articles in this series by Larson:
AI in biology: What difference did the rise of the machines make? AI works very well for proteins that lock into a single configuration, as many do. But intrinsically disordered ones don’t play by those rules. The resulting problems aren’t a temporary bug — they’re a basic limitation of training a machine learning model on a dataset where proteins always fold neatly.
AI in biology: So is this the end of the experiment? No. But a continuing challenge is that many of the most biologically important proteins don’t adopt a single stable structure. Their functions depend on structural fluidity The core issue AI isn’t just missing data — AlphaFold’s entire approach is built on assumptions that don’t apply to disordered proteins.
AI in biology: The disease connection — when proteins go wrong Some of the most crucial proteins for human health—the ones we need to understand most urgently—are the very ones that AI has the hardest time modeling. The issue is not simply that AI struggles with intrinsically disordered regions — it is that the very premise of IDR behavior contradicts the way these models operate. This isn’t just a flaw — it’s a fundamental crack in the foundation of AI’s “revolutionary” claims.
AI in biology: The future AI didn’t predict. It doesn’t look like the past. Physical systems that evolve over time but don’t follow a fixed formula have always presented a deep challenge to AI. The problem of outliers or “edge cases” has frustrated AI scientists and engineers (and now structural biologists) for decades, and there’s no good answer yet.