
AI in Biology: What Difference Did the Rise of the Machines Make?
AI works very well for proteins that lock into a single configuration, as many do. But intrinsically disordered ones don’t play by those rules(This is the second part in Erik J. Larson’s series on the attempt to understand protein folding using AlphaFold. The first part is here.)
Rise of the Machines
AI is a relative latecomer to the challenge of predicting the folding of proteins, though its arrival was perhaps inevitable. As with all machine learning methods, the challenge for AI involves “fitting” a candidate protein shape to a set of features, a problem duplicated over and over in the annals of predictive methods using machine learning.
AI has already proven its prowess in classification tasks—analyzing X-ray images to detect diseases like cancer, for instance, where the goal is a simple binary prediction: yes or no. Other machine learning models excel at time-series predictions, such as tracking heartbeats to detect subtle anomalies that might signal cardiovascular problems. Protein folding is yet another challenge — one that won’t require a revolutionary new approach but more like importing existing algorithms like deep neural networks to the new domain.
On cue, DeepMind’s Demis Hassabis and John Jumper entered the fray with the AlphaFold project—a name that AI enthusiasts will recognize as echoing AlphaGo and AlphaZero, the game-playing systems that dominated Go, chess, and other strategic challenges at superhuman levels. Protein folding is not a game, of course. But the AI playbook is largely the same.
Of course, even if the playbook is the same, the difference still matters. Next, we turn to how AlphaFold learns to predict protein folds — and when and why it doesn’t.
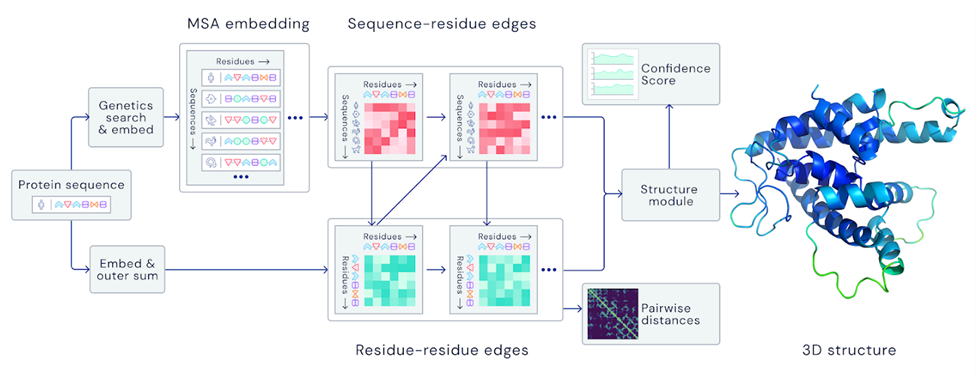
How AlphaFold learns — and why that matters
At its core, an AI approach to protein folding will mean engineering a machine learning system that can be trained to predict a protein’s three-dimensional shape from its amino acid sequence, as in the diagram above.
Enter AlphaFold. Instead of relying on direct experimental observation, it is trained on very large (read: huge) datasets of known protein structures, using machine learning to predict the shapes of previously unseen proteins. It does this by drawing on two key sources of information.
First, AlphaFold analyzes evolutionary history. By comparing families of related proteins across species, it identifies patterns in amino acid sequences that have remained conserved over millions of years. Proteins that share common ancestry often fold in similar ways, and AlphaFold exploits this fact, treating the past as a guide to the unknown.
Second, the system incorporates physico-chemical constraints — the fundamental principles that dictate how proteins fold. Hydrophobic regions tend to bury themselves inside the protein’s core, avoiding water. Hydrogen bonds stabilize helices and sheets. Steric hindrance prevents atoms from overlapping. AlphaFold does not merely rely on statistical pattern recognition; it refines its predictions by integrating these physical laws of molecular behavior.
The training process given these features is compute-intensive and requires high-powered processing on specialized hardware, such as Google’s TPUs or Nvidia’s GPUs. The system is trained on a database of known protein structures with the training features extracted, primarily from the Protein Data Bank (PDB). The PDB is a repository of experimentally solved structures, mostly obtained via X-ray crystallography and cryo-electron microscopy. Crucially, these methods work best on proteins that fold neatly into a stable, well-ordered shape. That means the dataset AlphaFold learns from is heavily biased toward structured proteins — proteins with a clear, single “ground truth” shape. But compared to traditional lab methods, the AI approach is dramatically faster and can generate useful predictions in hours rather than months or years of painstaking experimental work. The recipe for success is as follows.
How AlphaFold’s training process works
- Input Sequence – AlphaFold takes an amino acid sequence as input.
- Evolutionary Analysis – It compares the sequence against databases of known proteins, identifying patterns through multiple sequence alignments (MSAs).
- Structural Constraints – It includes biochemical and physical principles, such as hydrophobic interactions, hydrogen bonding, and steric hindrance.
- Deep Learning Prediction – Using deep neural networks, AlphaFold trains a predictive “model” of protein folding, and uses the model to generate a 3D structural rendition of a candidate protein by recognizing statistical relationships between sequence features and folded shapes.
- Confidence Scoring – Each region of the predicted structure is assigned a confidence score, indicating how reliable the prediction is. Since many proteins receive a high confidence score, the method is deemed a success — on the proteins it was trained on.
- Iterative Refinement – The model optimizes its predictions by minimizing errors, improving accuracy compared to previous iterations.
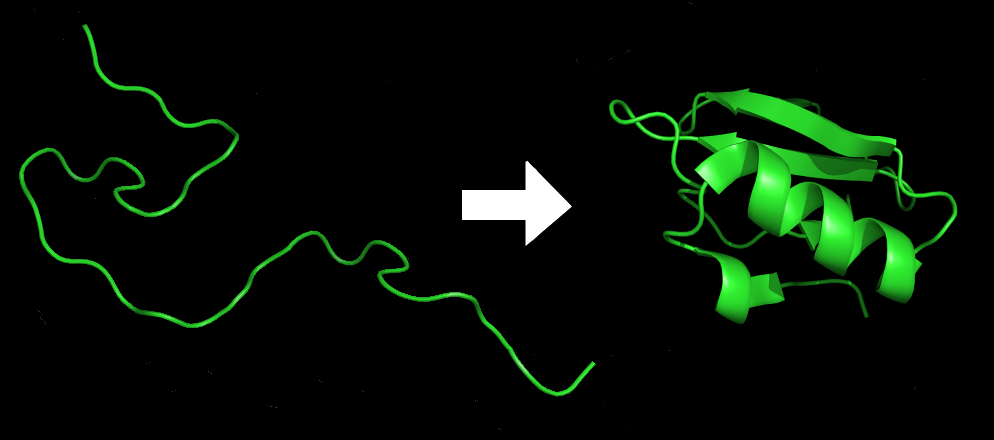
The process described here is essentially the way statistical machine learning works generally, since the beginnings of AI in the mid-20th century. And, it works astonishingly well. It works well, that is, for proteins that behave like static puzzles — those that lock into a single configuration, as many do. But intrinsic disorder, in this case “intrinsically disordered proteins (IDPs)” and “intrinsically disordered regions (IDRs),” don’t play by those rules. Unlike stable proteins, they exist in a constantly shifting ensemble of forms. Rather than having a single ground-truth structure, they have many fluctuating configurations based on environmental factors like temperature, pH, binding partners, and the effect of post-translational modifications (more on that later).
Because AlphaFold is designed to predict one best-fit structure, when encountering intrinsic disorder in proteins, it either outputs a low-confidence prediction or forces the IDP into an unrealistic stable form that it never actually adopts in nature. These outcomes show up in predictive results as very long confidence scores or just gibberish.
The hard fail on IDR and IDPs isn’t a temporary bug — it’s a fundamental limitation of training a machine learning model on a dataset that assumes proteins always fold neatly. This brings us back to AI’s past.
Note: Erik J. Larson writes the Substack Colligo.
Here’s the first article in this series by Larson: AI in biology: AI meets intrinsically disordered proteins. Protein folding — the process by which a protein arrives at its functional shape — is one of the most complex unsolved problems in biology. The mystery of protein folding remains unsolved because, as is so often the case with AI narratives, the reality is much more complicated than the hype.
Here are the rest: AI in biology: So is this the end of the experiment? No. But a continuing challenge is that many of the most biologically important proteins don’t adopt a single stable structure. Their functions depend on structural fluidity The core issue AI isn’t just missing data — AlphaFold’s entire approach is built on assumptions that don’t apply to disordered proteins.
AI in biology: The disease connection — when proteins go wrong Some of the most crucial proteins for human health—the ones we need to understand most urgently—are the very ones that AI has the hardest time modeling. The issue is not simply that AI struggles with intrinsically disordered regions — it is that the very premise of IDR behavior contradicts the way these models operate. This isn’t just a flaw — it’s a fundamental crack in the foundation of AI’s “revolutionary” claims.
AI in biology: The future AI didn’t predict. It doesn’t look like the past. Physical systems that evolve over time but don’t follow a fixed formula have always presented a deep challenge to AI. The problem of outliers or “edge cases” has frustrated AI scientists and engineers (and now structural biologists) for decades, and there’s no good answer yet.